
On Target?
Illustration by Gracia Lam
The promise and challenges of molecular therapies
Two paintings hang inside the office of Dr. David Andrews. They show patterns of dots. Some look like oversized peppercorns scattered on the canvas; others swirl together in cinnamon bun shapes. They catch the eye. If that eye is yours or mine, then the patterns probably won't hold any meaning. They're attractive (or not). To the eye of the indigenous artists in Australia who created them, however, they hold bags of meaning. "That pattern goes back about 6,000 years, and it depicts an area in the northwest of Australia where it gives the locations of safe places to camp and places to get water in an area that is inhabited by highly toxic lizards," he says, pointing to the swirliest of the two.
Andrews explains that the dots represent an aerial view of venom-secreting bushes in the desert that kill everything around them. "Because I'm not aboriginal, there's no way I could recognize where that is, but aboriginals know the location from that drawing. They know where that is," says the head of Biological Sciences at Sunnybrook Research Institute (SRI).
The meaning is there, then, for those in the know: the dots provide a kind of map for navigating through dangerous terrain; they mark where to go, if not exactly how to get there. It's a journey beset by pitfalls, but those who reach the destination are rewarded with safety, maybe even life.

Dr. David Andrews, shown with graduate student Elizabeth Osterlund, is working on a small molecule he thinks may have therapeutic potential. To help him and others find and validate such drug targets, he is building an image-based platform at Sunnybrook Research Institute that can knock down hundreds of genes of interest one at a time and analyze what happens—completely robotically.
Photo: Doug Nicholson
Molecularly targeted therapy is one of the most avidly pursued areas in medicine. At its most basic, it works by attacking a specific molecule or mechanism. This could be a gene mutation or it could be a biological process, like blood vessel growth. This is in contrast to therapies that work nonspecifically, like chemotherapy, which usually attacks DNA or structural components of cells. Targeted therapy seeks to replace today's dominant but variably effective treatments that fecklessly poison healthy as well as diseased cells, and in doing so not only manage disease better, but also banish the effects that are the dark companions of too many therapies.
How are we doing in terms of living up to this potential? We're making progress, but we have a long way to go. Unlike what the paintings in the office of Andrews portray, there is no map. Even if there were, there are no people who know how to read it, in part because the targets, found deep within our genome, are continually shifting (more on that later). And, the genome is a very big place.
Certainly, there are some spectacular successes. Many are in cancer, where the science of genetics is most advanced; we now know that cancer is not one disease defined by its site in the body, but many diseases more usefully defined by gene mutations.
There are also triumphs outside cancer, such as the discovery by Canadian researchers of numerous mutations in schizophrenia, which has jolted people into reimagining this brain affliction as several disorders. Similarly, a U.K.-based group has shown that type 1 diabetes is multifold. On the back of this, they have developed a test for newborns that determines if the babies have a mutation that causes one form of the disease. If they do, then they can be given an oral anti-diabetes drug, rather than insulin—a life-altering benefit.
Within cancer, trastuzumab, trade name Herceptin, is one of the best-known targeted therapies. It works by meddling with the signalling of the HER2 receptor, a protein that is overexpressed in the cancer cells of about 20% of women with breast cancer. Patients' tumours can be tested for HER2 expression; if positive, then they are candidates for Herceptin, which studies show reduces rates of recurrence, increases odds of survival and helps stop cancer from spreading.
Gleevec (generic name imatinib) is a small molecule inhibitor that has transformed the outlook for patients with chronic myeloid leukemia, a rare cancer that affects white blood cells. Patients given it early in their disease cycle have gone from a state where 30% would die within five years of diagnosis, to 95% living 25 years or more.
Dr. Arun Seth is hoping his research might lead to similar good news. A senior scientist at SRI, he has dedicated his career to untangling the molecular basis of breast and prostate cancer. Twenty years ago, while at the National Institutes of Health in the U.S., he was the first to clone insulin-like growth factor binding protein 7, or IGFBP7, from breast cancer cells, showing that it played a role in this disease. Then about 10 years ago, now at SRI, his lab discovered that the production of IGFBP7 falls as the severity of breast cancer goes up: the more invasive the cancer, the less this protein is expressed. They began to study it even more intently.
This protein is one of the insulin-like growth factor (IGF) binding proteins that occur naturally in our bodies. As a class, binding proteins are part of healthy cell regulation. As their name suggests, they attach to growth factors to keep them away from their receptors, unless otherwise indicated. "They release growth factors only at those times when the receptor needs to be activated, so they are keeping growth factors and receptor signalling in check most of the time," says Seth.
Different growth factors have different specific roles, but as a whole, managing cell growth is their thing. How they do this is known to all biologists: they bind to IGF receptors, which in turn promote cell survival and trigger cell growth. When it's part of normal regulation, it's all good. Sometimes, though, it's not. Unchecked cell proliferation is a calling card of cancer, and scientists have long known that activation of the IGF1 receptor spurs growth of breast, prostate and other cancers.
It is against this backdrop that Seth's recent finding on IGFBP7, published by the high-status journal Science Signaling in late 2012, was a landmark. "The real function of this protein wasn't known until we published that Science paper. We discovered that it directly binds to the IGF1 receptor," says Seth. "It was a totally new, totally unexpected, finding."
In bypassing the growth factor and binding to the receptor, the protein cuts out a step to assert itself at the source, gumming up the receptor and thus stopping cells from growing or even surviving. Pinpointing this mechanism—and upending everything that was known about how it works—was a crucial bit of knowledge. More tangibly, it shows how IGFBP7 could be used as a therapeutic in tumours that need IGF1 to thrive.
In a parallel study to evaluate this protein's therapy potential, Seth's lab grew human breast tumour cell lines, and then put them into mice, where they formed tumours. He injected IGFBP7 into some of the mice; others served as "untreated" controls. A few weeks later, they found the protein had suppressed tumour formation, big time.
"We could see the tumours where we injected the protein were 10 or 20 times smaller than those in the control group. When we analyzed the tumours, we could show that those tiny tumours have huge amounts of IGFBP7 in them—so [the protein] was actually inhibiting tumour cell growth," says Seth. They also determined that it was kick-starting apoptosis, or programmed cell death, and subduing the cells. "When it binds to the cells, the cells just sit there. They don't grow. They senesce. And when you remove IGFBP7, then they grow," he says.
His lab is now doing next-generation sequencing on the cell lines he used in that study. Sequencing gives information about differences in the genetic material of what is being analyzed—a molecular signature of sorts. It has been instrumental in targeted therapy success stories, because it enables people to identify biomarkers that can then help with diagnosis, guide the choice of treatment and predict outcome.

Dr. Arun Seth has discovered a protein that suppresses the formation of tumours in breast cancer cells. The protein, IGFBP7, could be a future targeted therapy, but there are many questions still to answer.
Photo: Curtis Lantinga
Seth's use of it has led to discovering that not all types of the tumours they're studying respond to the protein. "What we found is that IGFBP7 is not going to cure or inhibit all types of breast cancer, only certain types." One of these is triple-negative breast cancer, so-called because it is driven neither by overzealous hormones—namely, estrogen or progesterone—nor by the HER2 receptor; thus, it cannot be treated with therapies that target hormone receptors or production, or HER2.
"We got lucky with IGFBP7, and we showed in another paper that the triple-negative cell line is completely inhibited with IGFBP7," says Seth. Given how invasive this tumour type is, it's an exciting discovery.
All of this bolsters the case for IGFBP7 as a future targeted therapy, but although he is hopeful, Seth hasn't uncorked the champagne yet, as he sometimes does when he gets a big grant or publishes a meaningful paper to celebrate with his lab. There are still questions to answer and issues to surmount.
"I would say drug resistance and metastasis are the two biggest things that limit the success we've been able to make in treating advanced disease," says Dr. Bob Kerbel, senior scientist at SRI.
Metastasis is when cancer spreads from its origin to distant places in the body, typically to the lungs, liver, brain and bones. If a primary tumour is "localized," or hasn't spread, then the outlook is often good. "A scalpel blade will cure the patient, so to speak. You remove the tumour and they're sometimes cured," Kerbel says. Or, if there's "minimal residual disease," microscopic tumours left behind after removal of the primary tumour, then treatment usually works. To wit, the five-year survival rate for skin melanoma is 98%. Once it has spread, that rate plummets to 16%. Prostate cancer has similar numbers: a nearly 100% five-year survival rate is smashed to 28% if the cancer has wandered. Metastasis isn't uncommon. In breast cancer alone, about 30% of women will develop it.
Genomic instability is the hallmark of cancer.
Added to this mess is resistance, when drugs stop working. Kerbel explains: "When chemotherapy was initially developed in Boston, for example, for cancer in children, there were some fantastic results right away: tumours would melt that previously had been untreatable. "So that was a breakthrough, but what was then noted is that in a high proportion of cases, even though the children lived longer, the tumours would recur and no longer be responsive to the same therapy. They became resistant," he says.
Kerbel says that for all their potential, even the newest targeted therapies—65 years from when chemotherapy was first given to those children with leukemia—can't dodge the issue of resistance. It's almost all down to our genome, that massive, complex and evolutionarily canny collection of genetic material that defines us as individuals.
"Cancer cells have this phenomenal, frightening ability to become resistant. Just as you can find so-called 'driver' mutations in a cancer cell that might lead to a new drug targeting the products of such mutations, like the gene being amplified for HER2, which is a good thing, that same process of mutation can do something you don't want: a cancer cell might mutate in such a way that a clone from that cell arises that is no longer sensitive to the drug.
"When you analyze what happened, it turns out that in some cases the mutant protein has mutated again, the mutant gene has mutated again," he says, incredulously. "Now, as a result of that new mutation, the drug is prevented from doing what it was designed to do."
Adds Andrews, "Genomic instability is the hallmark of cancer. By the time you have a tumour large enough to detect, you have millions of phenotypes, so what you're doing isn't necessarily evolving, but selective, so if you kill 99.9% of the tumour, there's still lots and lots left, and if those [cells] are resistant because their genetic makeup is different and doesn't fit the biomarker for the targeted therapy, then you won't kill those [cells]."
This wily capacity of cancer to evade defeat is a formidable challenge and part of the rationale behind a way of delivering therapy that Kerbel has pioneered for almost one-half of his 30 years as a cancer biologist. Called "metronomic chemotherapy," it entails drugs administered in low doses over long periods without prolonged breaks. It works in part by halting new blood vessel growth, a process called angiogenesis. Kerbel is investigating its use alone as maintenance therapy, and paired with drugs that target blood vessel growth, like Avastin (bevacizumab), which works by blocking vascular endothelial growth factor, or VEGF. In both cases, the idea is to sever a tumour's lifeline, its blood supply, over a lengthy, vacation-free period as a way to make drugs less noxious, thwart resistance and, one hopes, improve patient survival.
Avastin is approved for advanced forms of colorectal, kidney, lung and brain cancer in North America. In Europe and Asia, it's also approved for breast and ovarian cancer. A clinical trial of cervical cancer just ended with good results, and hundreds more trials are ongoing. "Avastin is turning out to be one of the most wide-spectrum cancer drugs on the market. If you look at the number of the types of cancers it's been approved for, it's impressive," says Kerbel, though he also notes the drug is expensive and can have undesirable side effects.
The biggest qualifier? "If there's an overall survival advantage, it might be no longer than two to five months, on average. Now, some patients will do extremely well, living for years longer, and some not at all," says Kerbel.
Again—resistance. "What can happen is VEGF may be the major driver of tumour angiogenesis in a patient's cancer, so you give the drug with chemotherapy, and it works, and the tumour changes, and it says, 'OK; we're going to make something else that drives angiogenesis,' because VEGF is not the only factor that drives angiogenesis. There are likely dozens!"
It also underlines that most targeted therapies need a sidekick, standard chemotherapy, to work. Kerbel calls this "one of the great, ironic things" about targeted therapy. "It was designed with the expectation it would replace bad, old, not-so-targeted chemotherapy. In the main, it has not. There are some exceptions, but a lot of the new targeted drugs, like Avastin and Herceptin, they're normally used initially with chemotherapy, and if you don't use that combination, the targeted drugs don't work well at all," he says—which returns us to the way the therapy is delivered and the case for metronomic chemotherapy.
"We're arguing that the nature of the chemotherapy partner, the backbone—what the drugs are and how they are administered—may have an impact on how well the combination of chemotherapy plus the targeted therapy works," says Kerbel. The metronomic chemotherapy concept is being tested in about nine Phase 3 clinical trials. Results from one such trial were announced at the 2013 American Society of Clinical Oncology annual meeting. The Dutch study showed that metastatic colorectal cancer patients benefited from receiving long-term maintenance therapy comprised of continuous, low-dose, daily metronomic therapy plus Avastin after receiving conventional higher-dose chemotherapy and Avastin.
You need to develop the biomarker together with the targeted therapy.
Clinical trials are an integral part of the equation. That they're problematic isn't just because they're expensive—though they are—or because they don't get enough funding—though they don't—but because their design is struggling to keep up with genomics advances.
Science tells us that disease—breast cancer, say—is many diseases. Even triple-negative breast cancer, which accounts for 15% of all breast cancer at most, has at least six genetic subtypes. Thus, the old model of enrolling 5,000 women with breast cancer in a trial to test a targeted drug doesn't cut it. Odds are, the drug won't show an effect, even if one exists.
"Statistically, if all breast cancer patients had been recruited into the Phase 3 clinical trial of Herceptin, it would have failed, because only 15% have the target, and of those 15%, only half of them respond. That would mean out of 100 patients, in a nonselective trial, roughly 10% would benefit from the drug. The other 90% wouldn't," says Kerbel.
Science also suggests that most targeted therapies will work best in combination, so it makes some sense to test them this way, which is not as simple as it sounds, for a host of reasons. Adding to their complexity won't help trials get cheaper or better funded. Building smarter, more flexible trials is essential, however, to building better therapies. Using biomarkers to enrol patients on the basis of biomarker status, and then tracking them throughout, analyzing who does and does not respond, will help greatly. Sophisticated genomics tools are increasingly enabling researchers to do this.
"You need to develop the biomarker together with the targeted therapy, so that when you do your clinical trial, you're already in a specific subset of patients, and in that case if you have the biomarker, you may be doing the clinical trial with the conventional therapy that they're already getting, and saying, 'Now can we add this to this very specific cohort as opposed to the others and see if we can see a benefit?'," says Andrews.
Better preclinical models are also part of the solution.
Scientists use animal models to test promising drugs. With these results, companies decide if they will move a drug into patient trials. They are, therefore, vital. The problem is, many drugs shown to work in preclinical or small, early-stage trials fail during larger, later-stage trials, says Kerbel. "How come the mouse models overpredicted what happened in the clinic? And how come the early Phase 2 clinical trials looked like they might be encouraging, and then you go to the large Phase 3 randomized trial and [the drug] bombs?"
Part of the problem, he says, is that much preclinical research is on localized, primary tumours, not metastatic disease, which is far more difficult to treat successfully, and despite that most patient trials enrol people with advanced cancer. He started a program at SRI to develop models with metastatic cancer, and he tests therapies in these, thus mirroring the conditions of clinical trials and real life. He knows most drugs they test won't work, because the disease is so advanced. They look for exceptions, and when they find them, they connect with oncologists at Sunnybrook and elsewhere to begin the process of testing them. He also shares his models with other researchers all over the world.
A recent paper from Kerbel's lab shows the fascinating implications of this focus. In the study, they redid the preclinical experiments of the drug Sutent, an antiangiogenic small molecule approved for kidney cancer. It was once touted as the next big thing for breast cancer after its preclinical tests showed a robust benefit for it alone or plus standard chemotherapy, which helped convince the drug maker to fast-track it into randomized controlled trials—not one, but four "gold standard" multimillion-dollar trials.
"They all failed. Every trial failed, and the drug has been dropped from clinical development in breast cancer," exclaims Kerbel. "For me, that is an example of one of the most compelling examples of something that worked in mice, but didn't work in the clinic."
Kerbel's lab reproduced the original methods that led to Sutent's clinical testing, but also added a metastatic model of breast cancer. We bow heads over the paper as he explains the graphs depicting the results. The lines show clearly what the original experiments on primary tumours found: the drug seems to work. It shrinks tumour volume, and the mice live longer. It's striking. The graph for the metastatic model, however, tells a different story: the mice don't live longer. The lines drop off. The drug doesn't work. "It bombed," says Kerbel. "Basically, we recapitulated what happened in the clinic.
"It's only one model, but if we're right, the implications are huge, because we're saying you'll get a better idea of the effect of targeted therapies or for that matter chemotherapy, or when combining them, when you replicate the circumstance of metastatic disease, because all Phase 1 trials deal with such bad disease, virtually all Phase 2 trials deal with bad disease and most Phase 3 trials are also advanced, metastatic disease trials."
As important as improving preclinical models and trial design are, they don't get to the why of metastasis or resistance, and how to prevent them—or how to find better targets. For that, more precise tools are needed. That's where Andrews connects with Kerbel and Seth, as well as other scientists at SRI.
Andrews is building a platform that will enable him and others to identify new targets to treat disease and associated biological processes. At its heart is the most advanced fully automated high-content screening microscope in the world, the FLIM Opera, which Andrews built. It is the only tool that can measure quantitatively in live cells proteins interacting with each other, which gives insights not possible with other technologies, such as if a drug is working.
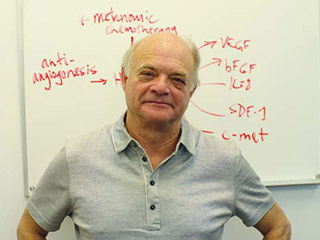
Dr. Bob Kerbel is a pioneer of metronomic chemotherapy, the administration of low doses of drugs given over a long period without prolonged breaks. It is here he places his hope, imagining a time when cancer may not be cured, but manageable over the long haul, with good quality of life.
Photo: Curtis Lantinga
Paired with this are custom-arrayed RNA-interference lentiviral libraries—basically a massive collection of genomic material. They offer a way to tailor a gene-finding mission, robotically. "For each scientist, we can assemble a custom library to knock down all of the genes that are relevant to the biology that they're studying," explains Andrews. Together with powerful image analysis software that his lab has developed, the platform is as close as one can get to a genome navigation tool for target discovery.
Take Kerbel's research on brain metastasis. As Andrews explains, Kerbel has developed a model of brain metastasis where 150 genes have been found as having changed in morphing from a pre- to post-metastasis state. "The infrastructure will allow us to knock out all of those genes one at a time, or in some limited combinations, and then using a cell culture model ask which ones of those genes have an effect on the cells, because you can't afford to test 150 genes one at a time in the mouse model to look for metastasis.
"Then you can ask, of those 150 changes, which are crucial for getting that metastatic phenotype? Those are your drug targets. Once you know which ones are required, then you can say, what's known about these? Which ones are the best targets? Then you can try and identify a small molecule that will inhibit that protein and then demonstrate to pharma whether you've actually identified a druggable target."
Andrews will use the "knockdown technology" to advance his work on interrupting protein-protein interactions as a way to reinstate cells' natural—but in cancer, faulty—impulse to commit suicide via apoptosis. He's identified a small molecule that he thinks has great cell-killing potential, but the class of proteins to which it belongs has a tendency to overexpress another protein that could make the cancer cells resistant to the small molecule. "It's what comes up with most targeted therapies: there's lots of ways around apoptosis, and tumours use most of them," he says. He will use the technology to see what's going on, and then find a compound to suppress that protein.
"It's very impressive," says Seth, who will use the system to watch how IGFBP7 does its thing in real time. He'll then combine these data with those from his next-generation sequencing of patient samples to distinguish which tumours will and will not shrink in response to the treatment, and from there figure out with which other compounds to pair the therapy, to be able to target tumour types defined by their molecular profile, in what he insists is the only way forward: "a multi-pronged approach."
On this, everyone seems to agree. First, targeted therapies will work best as part of a cocktail. With few exceptions, they will not be super drugs, able to take out disease with one perfect hit. Even before then, though, the processes of diagnosing, treating and tracking the outcome of disease need to be integrated, to use the knowledge streaming out of genomics and related fields. Globally, more than 70 research organizations (including Sunnybrook) in 40 countries have partnered to share genomic and clinical data. This is certain to accelerate the pace.
The goal is to achieve a standard of care when patients seeking treatment for any disease will have their genome sequenced toward delivery of much more precise treatment. Results would guide which targeted drugs to give, to ensure the best therapeutic response with the least toxicity. Recovery would be monitored biologically, allowing for therapy changes as needed, for example, if resistance emerges. Patients' lives would be drastically prolonged, and in some cases saved; quality of life, enriched.
An ambitious journey, to be sure. Are we on course? "I'm always optimistic," says Andrews.
Andrews' research is funded by the Canadian Cancer Society Research Institute (CCSRI), Canadian Institutes of Health Research (CIHR) and Ontario Institute for Cancer Research (OICR). Infrastructure support comes from the Canada Foundation for Innovation (CFI) and Ontario Ministry of Research and Innovation (MRI).
Kerbel's research is funded by the Canadian Breast Cancer Foundation (Ontario), CCSRI, CIHR, National Institutes of Health (NIH) and OICR. He holds the Canada Research Chair in Tumour Biology, Angiogenesis and Antiangiogenic Therapy.
Seth's research on targets in breast cancer is funded by the Canadian Breast Cancer Research Alliance, CIHR, National Cancer Institute of Canada and NIH. Infrastructure support comes from the CFI and MRI.
Related content
 Targeted therapy for lung cancer helps stem progression and is easier to tolerate: Research leads to new guidelines and moves therapy closer to being funded (from 2016 SRI Magazine)
Targeted therapy for lung cancer helps stem progression and is easier to tolerate: Research leads to new guidelines and moves therapy closer to being funded (from 2016 SRI Magazine)




